This article was originally published in 2023, and has been updated as of Mar. 2026.
If an FDA investigator walks into your facility and spots a product on hold for an out-of-range result, the first thing they'll want to know is whether your team is already investigating it. Not whether the hold is documented. Whether there's active investigation underway, a scope assessment has been run, and someone owns a corrective action.
That distinction, between a program that's documented and a program that's working, is what separates facilities that receive Form 483 observations from those that don't. FDA investigators aren't looking for a binder. They're looking for evidence that your team knows what's open, what the root cause was, what you're doing about it, and what you're doing to make sure it doesn't happen again.
If you can't walk them through that without scrambling, your CAPA program is a liability and not an asset.
What Auditors Look for When They Walk Your Facility
When an auditor walks your facility, they're trying to answer one question: do you actually have control of your food safety program? The answer comes through documentation, and not summaries or estimates, but start-to-finish records of what happened, what you found, and what you did about it.
A CAPA program has two sides: corrective actions that address problems after they occur, and preventive actions that stop recurrence across your operation. Most manufacturers have the corrective side reasonably well-covered. They fix the immediate problem, close the ticket, move on. The preventive side is where programs typically fall apart, and it's exactly what auditors are trained to look for.
Why Every Investigation Needs a Root Cause
The most common CAPA failure is treating each complaint as a one-time event. Fix the problem in front of you, close the record, done. The issue is that without understanding why it happened, the same problem reappears for a different customer, a different product, or at a different facility.
Repeat observations of the same issue are a recognized pattern that escalates investigator scrutiny, and in serious cases, triggers escalated enforcement action. If an investigator sees that pattern across your records, that's a very different conversation than a single contained incident.
Good preventive action starts with a question that most teams skip: "Could this happen elsewhere?" Running a scope assessment means more than closing the immediate event — it means examining your full program for related vulnerabilities before a second incident forces your hand.
Build Root Cause Analysis Into Every Investigation
Root Cause Analysis shouldn't be reserved for serious incidents. It should be standard practice from the first investigation. Finding the actual reason an event occurred, and not just the surface-level trigger, is what allows you to correct it properly and prevent recurrence.
CAPA can originate from anywhere: a customer complaint, an internal audit finding, a CCP deviation, an environmental positive, or a supplier non-conformance. Each distinct root cause, regardless of source, warrants its own corrective and preventive action. The most common mistake is confusing contributing factors with root causes when the real cause goes unaddressed, a single event can recur across multiple customers, lines, or sites.
Close the Loop: Effectiveness Verification Is Not Optional
Closing a CAPA record isn't the finish line. Two things still need to happen: follow-up with the affected customer to confirm the fix resolved their concern, and an effectiveness check to verify the corrective action actually worked and that the underlying issue hasn't resurfaced.
This last step is where many programs fall short, and it's a specific area of FDA scrutiny. A corrective action with no effectiveness verification is, from an investigator's perspective, an open loop; and open loops are what observations are made of.
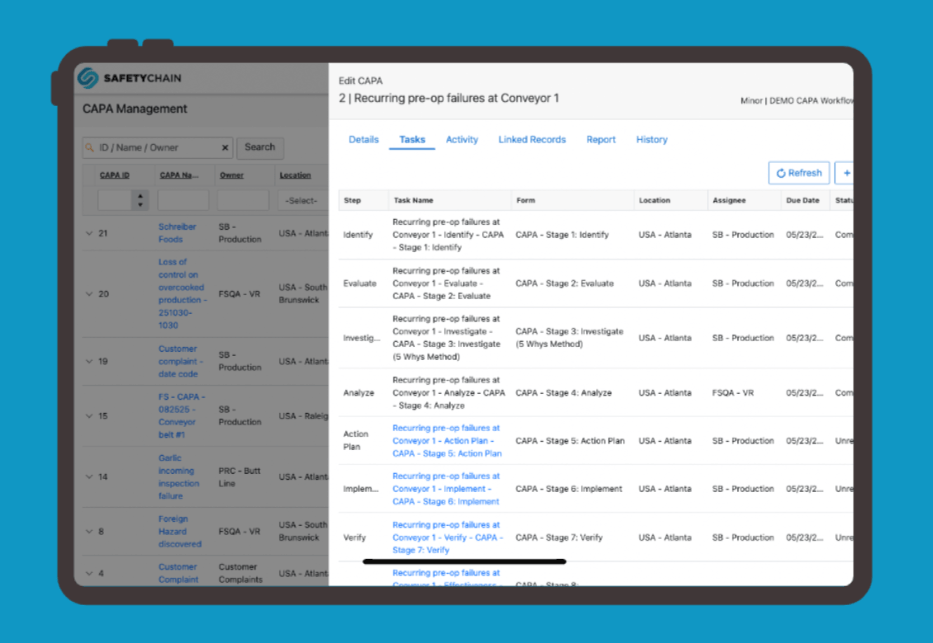
Document Everything Digitally and in One Place
The FDA doesn't want your best recollection. They want records organized, timestamped, and traceable, showing exactly what happened at every stage of an investigation. Paper files and spreadsheets don't hold up well under that scrutiny; when an investigator asks for everything associated with a specific event, the ability to produce it in minutes matters.
Solid recordkeeping delivers three things: a template for handling future incidents consistently, visible evidence that your program is active rather than theoretical, and reduced risk of follow-up action. A purpose-built digital CAPA system keeps all of it in one place, whether the action was triggered by a failed audit finding, an out-of-spec result, a temperature excursion, or a supplier deviation. Every record should be linked to its source event, every status change logged, and reports exportable on demand when an investigator asks.
What an Audit-Ready CAPA Program Actually Demonstrates
When an FDA investigator asks about your CAPA program, they want to see that your team knows what's currently open, what's been resolved, what the root cause was, and what's been done to prevent recurrence. That's hard to demonstrate when your records are spread across three systems and two filing cabinets.
An audit-ready CAPA program isn't defined by how many records you have; it's defined by how quickly and completely you can answer those four questions. If the answer requires pulling reports from multiple systems, chasing down paper files, or relying on someone's memory of what happened six months ago, the program isn't working the way investigators expect it to.
The facilities that walk away from FDA inspections without observations aren't necessarily the ones with the fewest problems. They're the ones that can demonstrate — clearly, completely, and immediately — that they know what happened, why it happened, and what they're doing to make sure it doesn't happen again.
Watch the FSMA Friday: Shoring Up Your Prevention Actions to Avoid FDA Form 483 replay: